
Carmen Ortiz-González, Laureano E. Carpio, Eva Serrano-Candelas, Rafael Gozalbes.
ProtoQSAR SL, c/ Nicolás Copérnico 6, Parque Tecnológico de Valencia, Paterna, Spain.
MolDrug AI Systems SL, Parque Tecnológico de Valencia, Paterna, Spain.
Peptides are molecules composed of 2 to 50 amino acids that can be used as drugs or drug candidates, as an alternative to small molecules due to some of their properties, such as their high degradation rate and the reduction of toxic metabolites release, which makes them an interesting option as therapeutic compounds.
In drug discovery field, in silico drug design has shown a notable progress in recent years, among which Quantitative Structure-Activity Relationship (QSAR) stands out. This method entails the development of statistical models from known data (using state of the art machine learning algorithms) so unknown molecular properties can be predicted. In silico methodologies supposes the reduction of the costs of research, as well as a faster collection of the results. Additionally, computational methods provide a more animal-friendly approach.
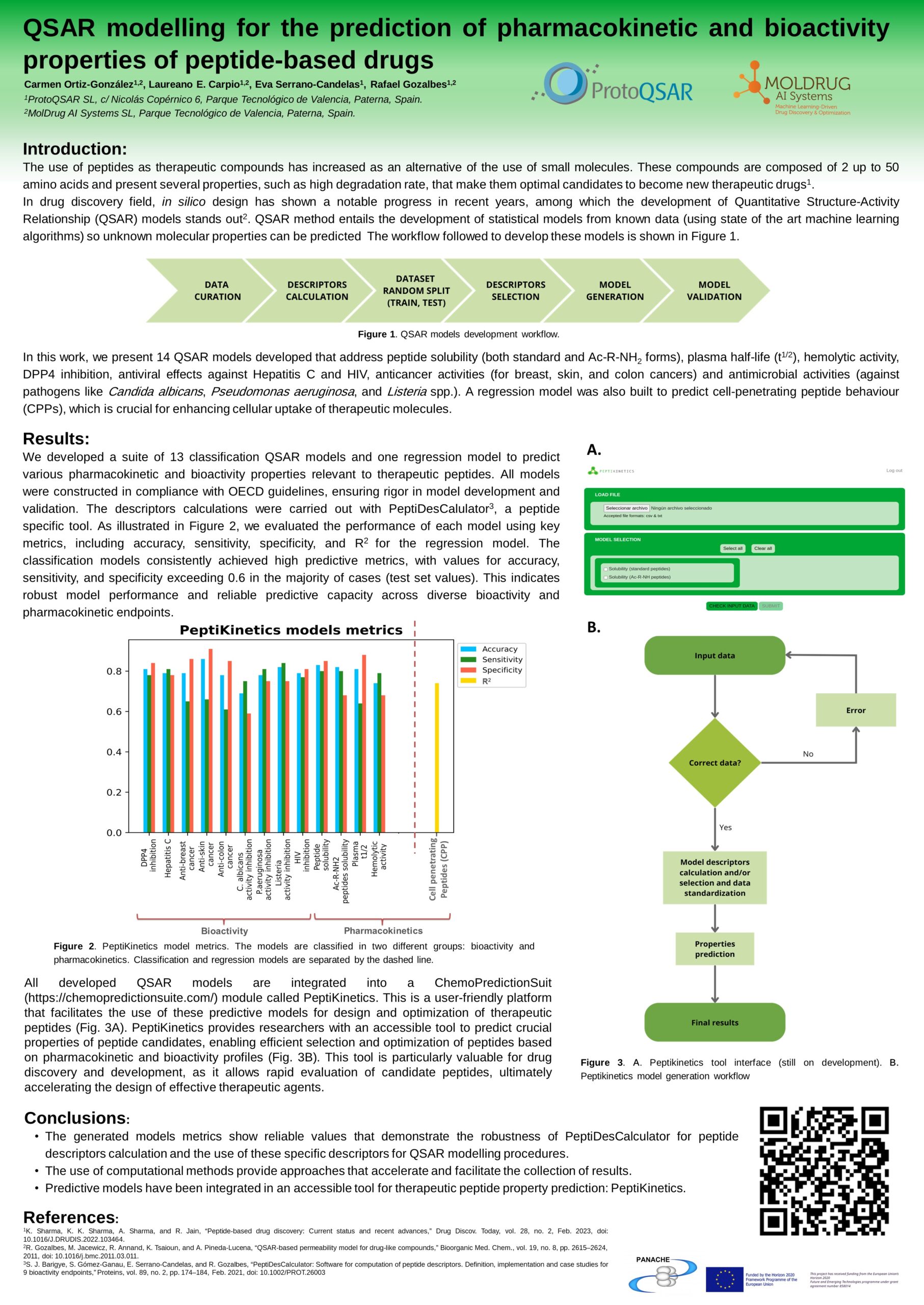
In this work, 14 QSAR models have been developed to predict peptide properties such as physicochemical parameters (i.e. solubility), bioactivity (i.e. inhibition of pathogen activity as C. albicans and P. aeruginosa), and pharmacokinetic (i.e. plasma half life). All of them suppose a wide range of tools that can enhance therapeutic peptides design.
These models have been developed following OECD standard procedures and an in-house specific software, PeptiDesCalculator, to calculate specific peptidic descriptors. Moreover, all these models were integrated in a computational tool called PeptiKinetics, which facilitates the use of these models for peptide design and optimization. All together form an accessible and easy way to develop and apply computational models for therapeutic peptides properties prediction.
cortiz@protoqsar.com

{kind=link}